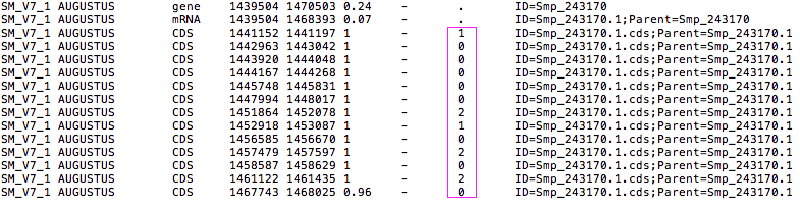
I had been trying to find a Perl or to write a Python script to extract protein sequences based on a gff annotation file. But just realised that both scrits don’t consider the 8th column, which indicates “phase”. As explained on the gmod website:
For features of type “CDS”, the phase indicates where the feature begins with reference to the reading frame. The phase is one of the integers 0, 1, or 2, indicating the number of bases that should be removed from the beginning of this feature to reach the first base of the next codon…For forward strand features, phase is counted from the start field. For reverse strand features, phase is counted from the end field.
This is particularly important for the first CDS feature, as it indicates whether to start translation from the first base or later, as shown in the title image. So need to modify the previous Python script to include this point (on both strands):
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
|
def pep_seq():
for t in db.features_of_type('mRNA', order_by='start'):
#print(t.sequence(myFasta))
print('>' + t.id)
seq_combined = ''
j = 0
for i in db.children(t, featuretype='CDS', order_by='start'):
j += 1
if j == 1:
pphase = i[7] # assign phase to the 8th column of first CDS
seq = i.sequence(myFasta, use_strand=True)
seq_combined += seq
seq_combined = Seq(seq_combined, generic_dna)
if t.strand == '-':
pphase = i[7] # assign phase to the 8th column of last CDS
seq_combined = seq_combined.reverse_complement()
if pphase == "0":
seq_transl = seq_combined.translate()
for i in range(0, len(seq_transl), 60):
print(seq_transl[i:i+60])
elif pphase == "1":
seq_transl = seq_combined[1:].translate()
for i in range(0, len(seq_transl), 60):
print(seq_transl[i:i+60])
elif pphase == "2":
seq_transl = seq_combined[2:].translate()
for i in range(0, len(seq_transl), 60):
print(seq_transl[i:i+60])
|
Full script is on my Github.