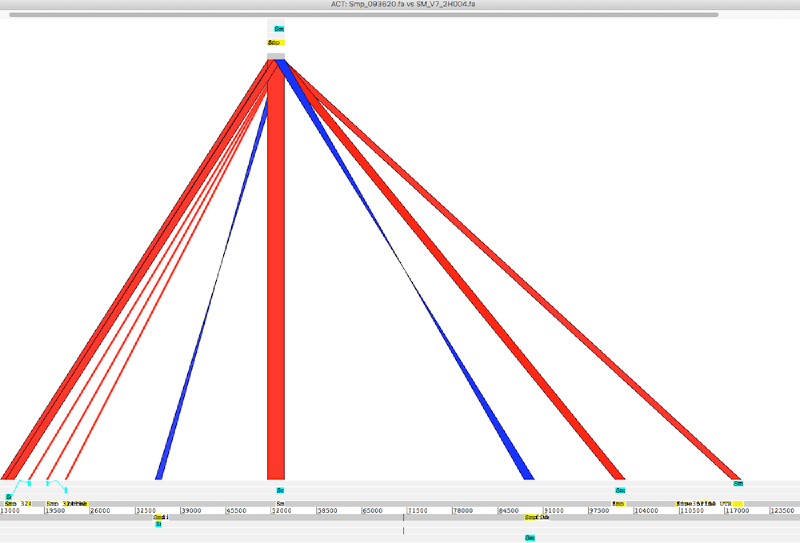
To use ACT we will need three essential files: sequence 1, sequence 2, and the comparison file (eg blast output). Additionally we can load annotation file to show features in the sequences (eg. gff or embl etc).
I my case I wanted to compare a gene or part of scaffold with a haplotype contig
gene to contig
I will need to
- extract gene sequence from gff
- compare gene sequence with contig sequence, ie. blastn
- modify the gene annotation to fit it’s sequence coordinate (eg. starting from 1)
- load gene seq (sequence 1), blastn output (comparison file), and contig seq (sequence 2) to ACT
- load gff annotations to sequence 1 and 2 to disply features
I have a bash script to do this (file_act_gene.sh). To load gff quicker the gff needs bgzip and tabix
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
|
#!/bin/bash
## usage: ./file_act_gene.sh [gene id] [contig No]
### get gene of target
grep $1 Sm_v7.2_Basic.gff > $1.gff
python ~/Google\ Drive/CodingPractice/py/gff2fasta.py $1.gff Smansoni_v7.fa gene > $1.fa
### blast gene with contig
blastn -query $1.fa -subject SM_V7_${2}.fa -outfmt 6 > $1-$2.blast.out
### annotation for contig
grep SM_V7_$2 Sm_v7.2_Basic.gff | sort -k1,1 -k4,4n > $2.gff
bgzip $2.gff
tabix $2.gff.gz
### annotation with relative coordinates
START="$(grep gene $1.gff|awk '{print $4}')"
cat $1.gff | awk '{print $1,$2,$3,$4-"'$START'"+1, $5-"'$START'"+1,$6,$7,$8,$9}'|tr ' ' '\t'> $1_rel.gff
rm -f $1.gff
### load into ACT: $1.fa, SM_V7_${2}.fa, $1-$2.blast.out
### read after: $1_rel.gff $2.gff.gz
|
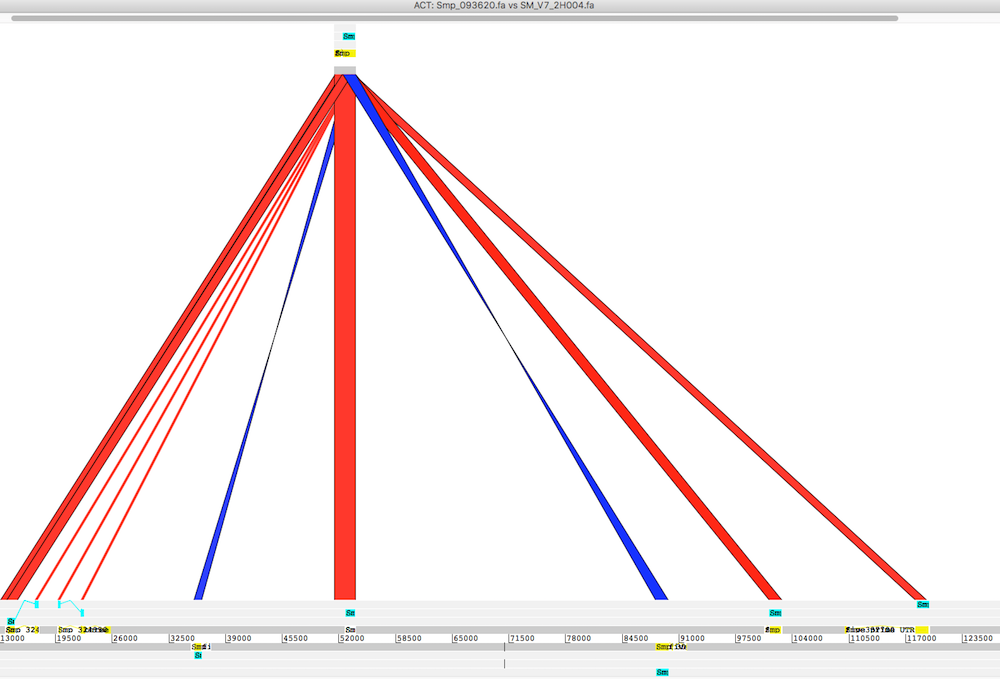
part of a scaffold to a contig
I need to supply the start and end coordinates of a target scaffold. To extract the sequence I used my colleague Alan’s python script:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
|
#!/bin/bash
## usage: ./file_act_coord.sh [scaffold No] [start Coord] [end Coord] [haplo contig No]
### get sequence of range
python alan_extract_fa.py Smansoni_v7.fa SM_V7_$1 $2 $3 > SM_V7_$1_$2-$3.fa
### blast fraction to contig
blastn -query SM_V7_$1_$2-$3.fa -subject SM_V7_${4}.fa -outfmt 6 > SM_V7_$1_$2-$3_$4.blast.out
### annotation for contig
grep SM_V7_$4 Sm_v7.2_Basic.gff | sort -k1,1 -k4,4n > $4.gff
bgzip $4.gff
tabix $4.gff.gz
### annotation for selected region
#### pay attention to usage of variable in awk
declare -i START=$2
declare -i END=$3
cat Sm_v7.2_Basic.gff | awk '$1=="SM_V7_""'$1'"' | awk '$4>='$START' && $5<='$END'' | awk '{print "SM_V7_""'$1'""_""'$2'""-""'$3'",$2,$3,$4-'$START'+1, $5-'$START'+1,$6,$7,$8,$9}'|awk '$4>0'|tr ' ' '\t' |sort -k1,1 -k4,4n> SM_V7_$1_$2-$3_rel.gff
### load into ACT: SM_V7_$1_$2-$3.fa, SM_V7_${4}.fa, SM_V7_$1_$2-$3_$4.blast.out
### read after: SM_V7_$1_$2-$3_rel.gff, $4.gff.gz
|
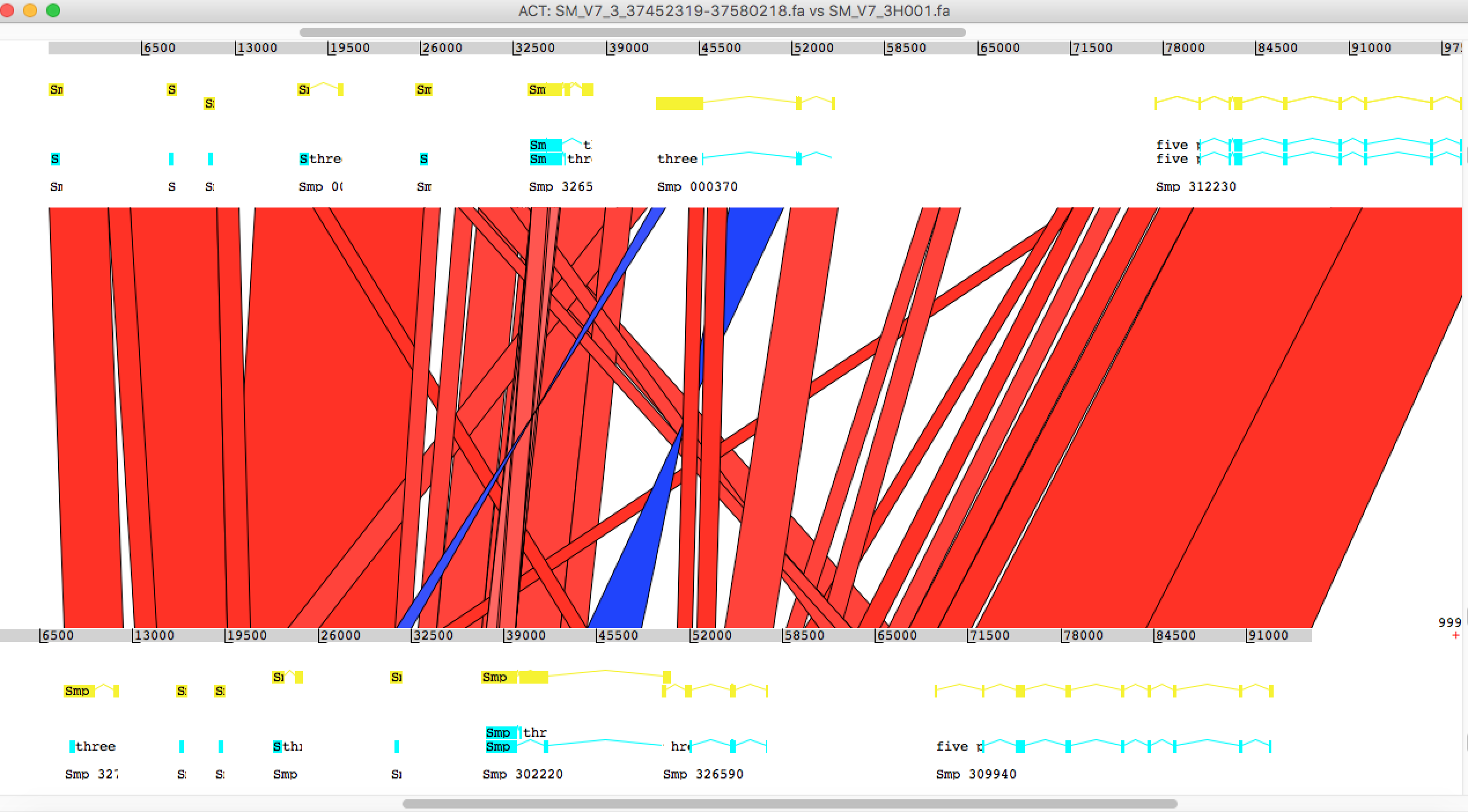
Just another tip for Artemis, after loading the fasta file, use the menu “Read Entry” instead of “Read Entry into” to load gff. The latter option can cause an error.