The input file is sorted by cluster name and start coordinate of the genes like this
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
|
chr id start end strand pfam
1 SM_V7_1 Smp_178420 48981354 48982520 - PF00001-7tm_1
2 SM_V7_1 Smp_319310 48986134 48988127 + PF00001-7tm_1
3 SM_V7_1 Smp_137300 48989538 48995030 - PF00001-7tm_1
4 SM_V7_1 Smp_137310 49003945 49005340 - PF00001-7tm_1
5 SM_V7_1 Smp_027940 49008050 49010380 - PF00001-7tm_1
6 SM_V7_1 Smp_137320 49025011 49029650 - PF00001-7tm_1
7 SM_V7_1 Smp_048050 65566436 65626137 + PF00011-HSP20
8 SM_V7_1 Smp_302270 65574596 65576347 + PF00011-HSP20
9 SM_V7_1 Smp_049230 65585301 65587122 - PF00011-HSP20
10 SM_V7_1 Smp_302280 65594848 65596602 - PF00011-HSP20
...
383 SM_V7_ZW Smp_137740 8280487 8305005 - PF17039-Glyco_tran_10_N
384 SM_V7_ZW Smp_336380 8321149 8334825 + PF17039-Glyco_tran_10_N
385 SM_V7_ZW Smp_142860 8337999 8348552 + PF17039-Glyco_tran_10_N
386 SM_V7_ZW Smp_306350 62981689 62988318 + PF17039-Glyco_tran_10_N
387 SM_V7_ZW Smp_333000 63016429 63024590 + PF17039-Glyco_tran_10_N
388 SM_V7_ZW Smp_099090 63053639 63061327 + PF17039-Glyco_tran_10_N
|
I have previously used a for loop to manually calculate the distances. After a bit of searching, there seems much easier solutions to calculate the distance (difference) between the end coordinate of gene A to the start coordinate of next gene within each pfam group
For instance using dplyr (lag function):
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
|
library(dplyr)
pfamCoord<-read.delim("cluster_geneCoord.txt", sep="\t", header=T)
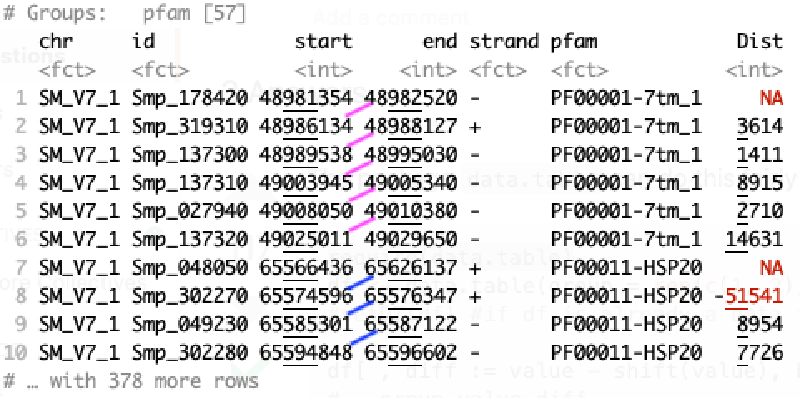
pfamCoord %>% group_by(pfam) %>% mutate(Dist = start - lag(end))
# A tibble: 388 x 7
# Groups: pfam [57]
chr id start end strand pfam Dist
<fct> <fct> <int> <int> <fct> <fct> <int>
1 SM_V7_1 Smp_178420 48981354 48982520 - PF00001-7tm_1 NA
2 SM_V7_1 Smp_319310 48986134 48988127 + PF00001-7tm_1 3614
3 SM_V7_1 Smp_137300 48989538 48995030 - PF00001-7tm_1 1411
4 SM_V7_1 Smp_137310 49003945 49005340 - PF00001-7tm_1 8915
5 SM_V7_1 Smp_027940 49008050 49010380 - PF00001-7tm_1 2710
6 SM_V7_1 Smp_137320 49025011 49029650 - PF00001-7tm_1 14631
7 SM_V7_1 Smp_048050 65566436 65626137 + PF00011-HSP20 NA
8 SM_V7_1 Smp_302270 65574596 65576347 + PF00011-HSP20 -51541
9 SM_V7_1 Smp_049230 65585301 65587122 - PF00011-HSP20 8954
10 SM_V7_1 Smp_302280 65594848 65596602 - PF00011-HSP20 7726
# … with 378 more rows
|
or using data.table (shift function)
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
|
library(data.table)
setDT(pfamCoord)
pfamCoord[, Dist := start - shift(end), by = pfam]
head(pfamCoord, 10)
chr id start end strand pfam Dist
1: SM_V7_1 Smp_178420 48981354 48982520 - PF00001-7tm_1 NA
2: SM_V7_1 Smp_319310 48986134 48988127 + PF00001-7tm_1 3614
3: SM_V7_1 Smp_137300 48989538 48995030 - PF00001-7tm_1 1411
4: SM_V7_1 Smp_137310 49003945 49005340 - PF00001-7tm_1 8915
5: SM_V7_1 Smp_027940 49008050 49010380 - PF00001-7tm_1 2710
6: SM_V7_1 Smp_137320 49025011 49029650 - PF00001-7tm_1 14631
7: SM_V7_1 Smp_048050 65566436 65626137 + PF00011-HSP20 NA
8: SM_V7_1 Smp_302270 65574596 65576347 + PF00011-HSP20 -51541
9: SM_V7_1 Smp_049230 65585301 65587122 - PF00011-HSP20 8954
10: SM_V7_1 Smp_302280 65594848 65596602 - PF00011-HSP20 7726
#setDF(pfamCoord) # convert back to old data.frame syntax
|
Finally we can summarise the differences (Dist) by group (pfam)
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
|
pfamCoord %>% group_by(pfam) %>% summarise(minDist=min(Dist, na.rm = TRUE), maxDist=max(Dist, na.rm = TRUE))
`summarise()` ungrouping output (override with `.groups` argument)
# A tibble: 57 x 3
pfam minDist maxDist
<fct> <int> <int>
1 PF00001-7tm_1 -14237049 37313397
2 PF00011-HSP20 -51541 54921
3 PF00012-HSP70 4828 4842
4 PF00014-Kunitz_BPTI 3703 46064
5 PF00026-Asp 11392 28849
6 PF00041-fn3 1212 1327
7 PF00079-Serpin 20471 45424
8 PF00089-Trypsin -3348130 138508
9 PF00096-zf-C2H2 5770 15632
10 PF00106-adh_short -43497156 53670503
# … with 47 more rows
|
or to summarise by pfam on each chr, because some pfam clusters exist on multiple chromosomes.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
|
pfamCoord %>% group_by(pfam, chr) %>% summarise(minDist=min(Dist, na.rm = TRUE), maxDist=max(Dist, na.rm = TRUE))
# A tibble: 67 x 4
# Groups: pfam [57]
pfam chr minDist maxDist
<fct> <fct> <int> <int>
1 PF00001-7tm_1 SM_V7_1 1411 14631
2 PF00001-7tm_1 SM_V7_3 -9539472 1870737
3 PF00001-7tm_1 SM_V7_4 -14237049 20454
4 PF00001-7tm_1 SM_V7_ZW -17590 37313397
5 PF00011-HSP20 SM_V7_1 -51541 54921
6 PF00012-HSP70 SM_V7_1 4828 4842
7 PF00014-Kunitz_BPTI SM_V7_2 3703 46064
8 PF00026-Asp SM_V7_3 11392 28849
9 PF00041-fn3 SM_V7_ZW 1212 1327
10 PF00079-Serpin SM_V7_6 20471 45424
# … with 57 more rows
|
Some distances are minus because genes are overlapped.