Transfer the old annotations to the new genome using RATT
RATT is a rapid annotation transfer tool for different genome Assembly versions, but can also be used for different Strains and even Species. And for each case therer is a preset parameter with different “l” (k-mer size), “c” (cluster size) and “g” (extending of cluster) values. It needs MUMer (nucmer) to run.
It takes about 11 hours for S. mansoni genome (~360Mb) annotation transfer and the resulting files are “*.final.embl”.
Then you can use a Perl script written by Avril Coglan at Sanger to convert the embl files to gff format. (BTW, you can really learn a lot about omics data processing from her blog)
After the run, RATT will give you a summary report in the output file, and below the line “CDS” you can numbers of gene models been transferred and not transferred. I got 95% transferred for the schisto transfer (Be aware that you might have one gene transferred to different locations: might be paralogs)
In addition you can evaluate it by compare the aa sequences of genes that you are confident in for the old assembely after transfer, using pairwise sequence alignment.
Gene finding in the new genome using Augustus / Braker
Augustus is a gene predictor for eukaryotic genomes, and can incorporate data from RNA-Seq. Thus, the predication accuracy can be greatly improved. It has currently been trained for various species on specific training sets.
The results are in GFF format.
Brief steps to run Augustus:
- bam2hints to product intron hints part
- bam2wig to product exon part; and wig2hints to convert wig to hints
- cat intron and exon hints and generate to the final hints file
Compare transferred gene models (RATT) and predicted models (Augustus)
Depending on the quality of current genome annotation, it is not always necessary to run both RATT and Augustus. For instance for a well-annotated genome, it is just sufficient to use RATT to transfer annotations. For a genome that is bad-annotated and not been used for many researchers, it is probably a good idea to take the Augustus gene set because it got trained from RNAseq evidence. But if you have done both and want to decide which to take, you can probably use following approaches:
- Load the annotations on a genome browser (e.g., Artemis, Apollo, and JBrowse (built with HTML5 and JavaScript!)) and evaluate the gene structure with RNAseq mappings.
- Compare the sequence similarities with those before transfer (better to have some golden standard genes as reference)
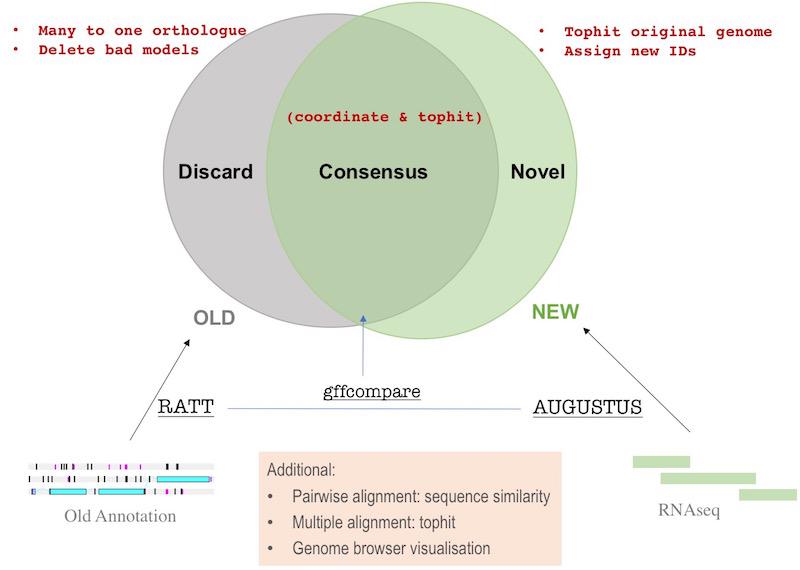
Finding consensus transcript using gffcompare
gffcompare was used to find consensus transcripts (and keeping the IDs) between RATT and Augustus gff files. The GFF file should contain only unique line before run gffcompare. I used RATT gff as a reference and compare Augustus one to that:
|
|
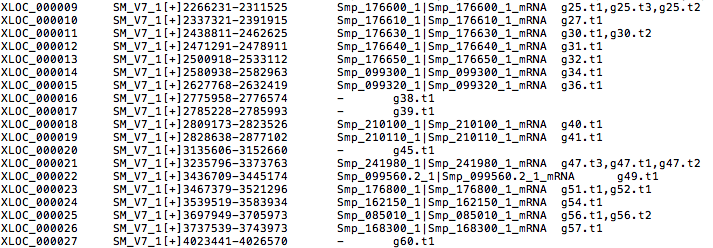
By doing this, I got among other files an annotated gtf (.annotated.gtf; labelling consensus ids from the reference on “transcript”), a .stats file, a .loci file containing all IDs in the same locus, and a detailed .tracking file.
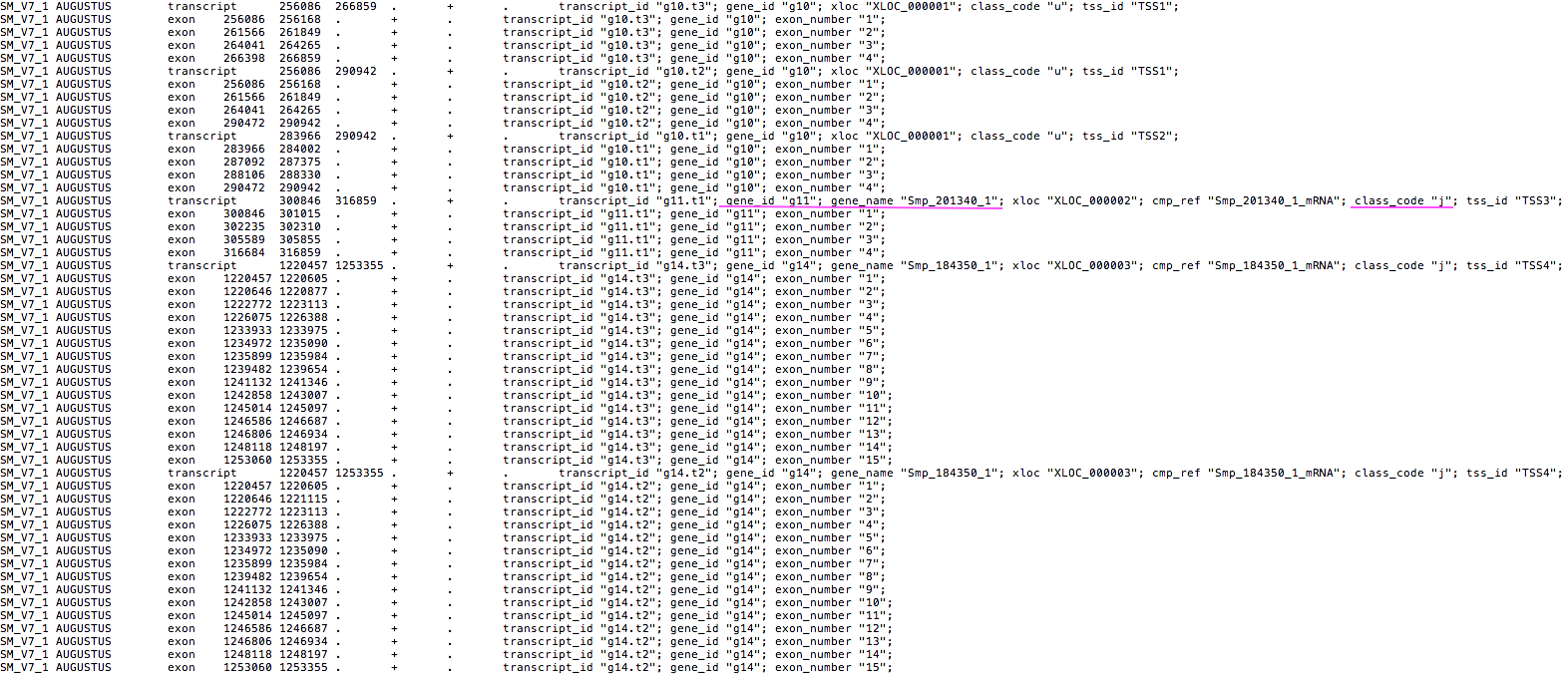
The .annotated.gtf file: (see larger image here)
{kind=link}
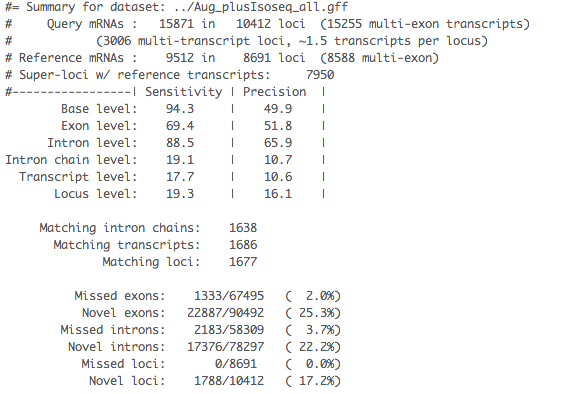
The .stats file:
The .loci file:
Note that in the gtf and tracking file there is a class_code with a character like “j” or “=” etc., telling you the difference between the two consensus transcripts. The code meaning is the same as from cuffcompare.
From this you can generate a list of consensus transcript IDs for later identifier transfer.
Confirming the consensus
Note that gffcompare define consensus based on their coordinates, so if one query gene is located in the UTR region of a reference gene, gffcompare will take the id from the reference, which might not be correct (they may hove no overlapping exons). So I performed sequence alignment for both gene sets, extract the tophit for each query gene, and compare it to gffcompare consensus id. If these two approaches point to the same reference ID, I will put it into my id pair list.
Pairwise alignment to detect sequence similarities
Although we got consensus transcript id pairs, we donot know to what extent the sequences are changed. So a pairwise sequence alignment (here is how to) will be helpful to evaluate this. We will be very confident in those having > 95% agreement in their sequence lengths and similarities.
|
|
Check for cases of gene merge and split
Merge: ONE Augustus to MANY Ratt genes:
|
|
Split: ONE Ratt to MANY Augustus
|
|
Assessment of non-consensus, untransferred and novel transcripts
Not transferred, or no-consensus from the OLD set
Because gffcompare only takes one reference ID from each Augustus transcript, so if there are splice variants in RATT, these will not be coded as “consensus”. We can manually check if they have isoforms in the ID pair list.
And for those not been transferred by RATT, we can align them to Augustus gene set and take thoes with 95% confidence.
For the rest, they might be bad gene models, psudo genes, or having a hugo change due to different genome assembly versions.
Novel transcripts in the NEW set
- Check whether we should keep them: are they short, or are they in the UTR region of another gene? Do they code for functional products (e.g., domains)?
- If we keep, give them new IDs.
Identifier transfer using string replacement
By doing above steps, we should be able to get a long list with id pairs from RATT and Augustus. We should check again for cases of merging and spliting, and give new identifiers if necessary.
For the ID transfer, I used a perl script for replacing strings from the ID pairs list.
Post-transfer assessment
To relieve the concers from the community about changes in their beloved genes, the following assessment would be helpful:
- Have the genes changed to different chromosomes?
- What are the sequence difference (esp. amino acid) for genes that people have studied and published?