Tags
14 pages
Bioinformatics
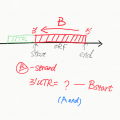
Get the UTR region for non-annotated transcripts
Remove all features from A that intersect/overlap with those in B
Installing genometools for MacOS
Meta-analysis of RNAseq studies
Get fasta sequences for features in a gff file using Python
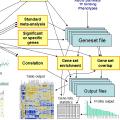
Genome annotation pipeline and tools
1
2
3