In the annotation we often have mRNA and CDS and often no UTR annotated. Therefore for genes
- on plus (+) strand without 3’UTR: mRNA end_coord = last CDS end_coord
- on minus (-) strand without 3’URT: mRNA start_coord = last CDS start_coord
We wanted to extract a pre-defined 3’UTR region (1kb downstream 3’end). Here is a workflow:
-
- check if there is any gene with an 3’UTR
-
- if no UTR, we can calculate the distance between transcript A end to the start of transcript/gene B (take only 1 transcript per gene), on each scaffold
-
- check the distance, if <= 1kb then take the distance; if >1kb then take 1kb. Modify the coordinates (pay attention to the strandness) and output to gff fomat
-
- extract sequences
1. To check if any gene has 3’UTR
The idea is to compare the coordinates between mRNA and the last CDS. Apollo always output CDS in their structural order, rather than coordinates, so the last CDS is always at the bottom. For other programs the CDS might be ordered by coordinates.
After grepping the transcript features, we can use the bash command awk 'NR==1; END{print}' to print first (mRNA) and last (last CDS) line, and compare the start/end coordinates (if uniq then gives 1, otherwise 2)
|
|
The output looks like this. We need to check if there is any transcript with 2 in their 2nd or 3rd column.
|
|
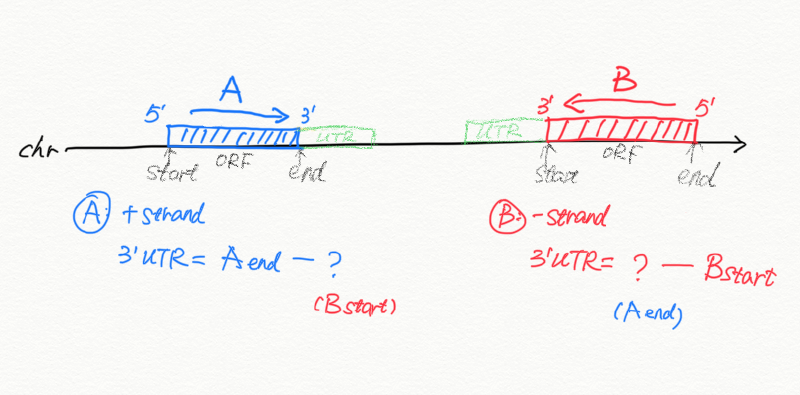
2. Calculating the distance between 3’end and next gene Start
In case of no UTR annotation for all genes, we first prepare a list of primary transcripts (.1), and order them by scaffolds and start coordinates. So they will appear adjacent.
- If gene A is on the + strand, we compare it to the next gene B
- if the next gene B is on the same scaffold, then we take distance as A_end to B_start
- if the next gene B is not on the same scaffold, then gene A is the last gene, we take A_end to scaffold_end.
- If gene A is on the - strand, we compare it to the previous gene C
- if the previous gene C is on the same scaffold, then we take distance as C_end to A_start
- if the previous gene C is not on the same scaffold, then gene A is the first gene, we take scaffold start (1) to A_start.
We can wrap it into R script:
|
|
Output gives the distances
|
|
- for + strand: col3 comes from mRNA 3’end, col4 comes from start_coord of the next gene
- for - strand: col3 comes from end_coord of the previous gene, col4 comes from the mRNA 3’end
3. Adjusting the distance/length
Now it is easy, if the distance is <=1kb then take the distance, if not then adjust the coordinates
|
|
for example:
|
|
now becomes
|
|
We can combine the files (part*.txt) and make a final gff. Then extract the sequences
|
|
Just to check the lengths of extracted 3’UTRs (show random 5 genes):
|
|